Understanding ISO 14971 and pFMEA: Regulatory Expectations in Medical Device Development 101

Navigating the complex waters of medical device development is akin to setting sail on an exciting but challenging voyage. The promise of innovation is tantalizing, but the need to adhere to stringent regulatory requirements and standards can feel like taming a wild sea. In this journey, having an experienced captain like Nectar Product Development, armed with ISO 13485:2016 as the navigational chart and FDA regulations as the guiding constellations, is essential. We seamlessly blend the winds of innovation with the North Star of compliance, ensuring a course that leads to success.
Key Regulatory Requirements in Medical Device Design and Development
In the realm of medical device design and development, regulatory requirements are akin to a labyrinth, with crucial benchmarks outlined in ISO 13485:2016 and FDA 21 CFR part 820.32 (c). Let’s delve deeper into these documents to understand their significance.
ISO 13485:2016 – Section 7.3.2
Section 7.3.2 of ISO 13485:2016 focuses on design and development within quality management systems. It encompasses several critical facets, including:
- Documentation of design and development stages: This involves meticulously documenting the various stages of the design and development process.
- Specification of review requirements: At each design stage, there’s a need to specify review requirements to ensure thorough evaluations.
- Definition of verification, validation, and design transfer activities: These activities must be defined for each development stage to ensure product quality and compliance.
- Allocation of responsibilities and authorities: Clear roles and responsibilities for design and development activities are crucial.
- Establishment of methods for traceability: Ensuring traceability from outputs to inputs is essential to maintain product integrity.
- Determination of necessary resources: This includes assessing personnel competency and ensuring that adequate resources are available.

It’s worth noting that Nectar Product Development is ISO 13485:2016 certified, underscoring our commitment to quality and compliance.
What are the Key Regulatory Requirements in Medical Device Design and Development?
In the realm of medical device design and development planning, there exists a labyrinth of regulatory prerequisites. The foundational benchmarks are succinctly delineated in section 7.3.2 of ISO 13485:2016 and FDA 21 CFR part 820.32 (c). Let’s delve deeper into these documents to unveil their quintessence.
FDA 21 CFR Part 820.30 – Design Controls and Preplanning
FDA 21 CFR Part 820.30 places a strong emphasis on design controls, which necessitate meticulous preplanning. This involves mapping out milestones, anticipating financial and technical challenges, and establishing a clear release timeline for your medical device. According to this regulation, every manufacturer must formulate and uphold plans that outline design and development activities, along with defined responsibilities for implementation. These plans should also encompass interactions with various groups or activities providing inputs to the design and development process, undergoing periodic review, updates, and approval as the development process progresses.
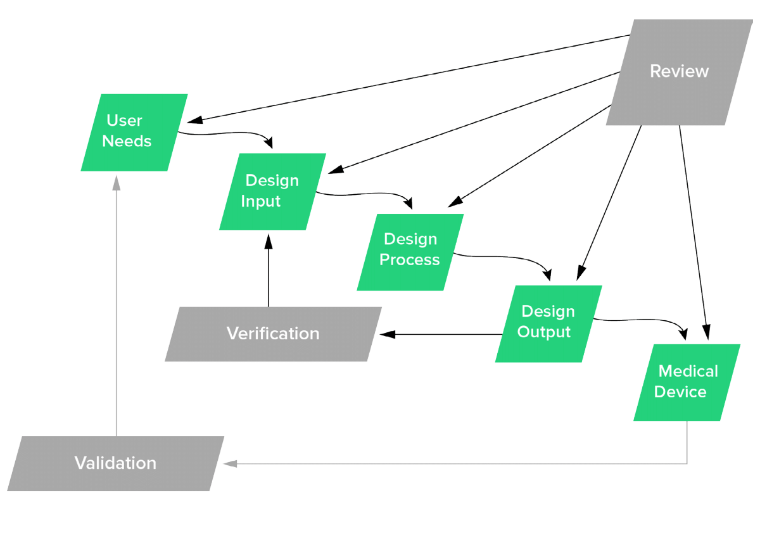
Infographic that shows the design control process in medical device development. It begins with user needs and ends in a medical device.
Why is Integrating Risk Management into the Design Plan Important According to ISO 13485:2016 and 21 CFR Part 820.30?
An indispensable takeaway is the integration of risk management activities within the design and development plan, as stipulated by both 21 CFR Part 820.30 and ISO 13485:2016 section 7.3.2. Crafting a design plan isn’t as simple as generating a Gantt chart; it necessitates thoughtful consideration. While the official guidelines do not explicitly demand anticipated delivery dates, they are commonly expected by manufacturers and businesses. The intricacies of these guidelines necessitate the inclusion of all projected project activities across groups and interactions, delineating core team competencies. Every developmental step should be meticulously documented, complying with design control processes.
What is a Quality Management System (QMS)?
A Quality Management System (QMS) is a structured framework implemented by organizations to ensure consistent quality in their products or services. It encompasses a set of processes, procedures, policies, and documentation that outline the methods and practices required to meet customer expectations, regulatory requirements, and industry standards. A well-designed QMS aims to enhance efficiency, minimize errors, and foster continuous improvement throughout the organization’s operations. By monitoring and controlling various aspects of production, delivery, and customer interactions, a QMS contributes to the overall reliability, safety, and satisfaction associated with the organization’s offerings.
How does ISO 13485:20106 QMS work?
ISO 13485:2016 serves as a comprehensive framework tailored to the unique demands of the medical device industry. This standard orchestrates processes throughout the product lifecycle, covering design, development, production, distribution, and post-market activities. It ensures organizations identify, evaluate, and mitigate potential risks while aligning product designs with intended functionality and regulatory prerequisites. Effective supplier management is also emphasized to ensure the integrity of components and materials, all while upholding quality, safety, and compliance in the medical device landscape.
The Significance of pFMEA
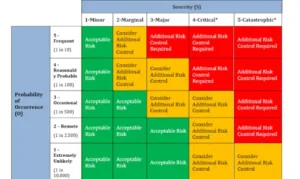
One of the key components in ensuring the safety and effectiveness of medical devices is the implementation of pFMEA, or Process Failure Modes and Effects Analysis. This systematic approach allows us to proactively identify potential failure modes within the manufacturing and design processes. By conducting pFMEA, we can assess the severity, occurrence, and detectability of these failure modes, ultimately prioritizing which areas require the most attention and mitigation efforts. This proactive approach aligns perfectly with our commitment to risk management, ensuring that every possible precaution is taken to guarantee the quality and reliability of our medical devices.
Embracing ISO 14971 for Risk Management
ISO 14971 is another critical standard that plays a pivotal role in the medical device development landscape. This standard specifically addresses risk management for medical devices, providing a structured framework for identifying, assessing, and controlling risks throughout the product’s lifecycle. At Nectar Product Development, we embrace ISO 14971 as an integral part of our risk management strategy. By aligning our processes with this standard, we ensure that risks are systematically evaluated, documented, and mitigated, resulting in safer and more reliable medical devices for the benefit of patients and healthcare providers.
ISO 14971 guides our team through a systematic approach to risk mitigation, enhancing our ability to deliver safer and more reliable medical devices. This approach involves a careful examination of potential hazards and their associated risks, considering factors such as severity, occurrence, and detectability. Each identified risk is meticulously documented, allowing for a clear and comprehensive understanding of the potential challenges that may arise during the device’s lifecycle. Moreover, ISO 14971 promotes proactive risk mitigation strategies, ensuring that every possible precaution is taken to guarantee the quality and safety of our medical devices.
The ultimate beneficiaries of our commitment to ISO 14971 are the patients and healthcare providers who rely on the medical devices we develop. By adhering to the stringent risk management practices outlined in this standard, we minimize the potential for adverse events and maximize the safety and effectiveness of our products. Patients can have confidence in the devices they use, knowing that extensive risk assessments and mitigation efforts have been diligently conducted. Healthcare providers, on the other hand, can trust in the reliability of the tools and equipment they employ in their critical work, allowing them to deliver the best possible care to their patients. At Nectar Product Development, ISO 14971 is not just a standard; it’s a commitment to excellence and safety in the world of medical device development.
Our Vision for the Future
As we move forward into the ever-evolving landscape of medical innovation, our vision at Nectar Product Development remains steadfast. We are committed to staying at the forefront of regulatory compliance, continuously integrating the latest standards and best practices into our processes. We understand that the journey of medical device development is not static; it’s a dynamic voyage filled with challenges and opportunities. With our experienced team, dedication to quality, and proactive approach to risk management, we are poised to navigate these waters with confidence. Our goal is to continue delivering innovative, safe, and compliant medical devices that make a meaningful impact on the healthcare industry and the lives of patients around the world.
In conclusion, successfully navigating the regulatory expectations in medical device development requires a meticulous approach that combines innovation with compliance. Nectar Product Development stands as a seasoned captain in this journey, guiding you through the intricacies of ISO 13485:2016 and FDA regulations. We integrate risk management into our design plans, ensuring comprehensive documentation and adherence to design control processes. Our commitment to a robust Quality Management System, aligned with ISO 13485:2016, underscores our dedication to delivering safe and compliant medical devices. As we sail into the future, embracing innovative technologies like ISO 14971 and pFMEA, Nectar Product Development is ready to face the challenges and opportunities that lie ahead in the ever-evolving world of medical innovation.